Summary
In this tutorial we will see examples on parseblast output when applying to a WU-BLAST file. This perl script is included in the gff2aplot distribution. There are three basic aligment formats that can be generated from a blast file by parseblast.pl, but all three must produce the same plots by gff2aplot. We will also see the raw output from gff2aplot and how to customize it a little bit.
NOTE.- For the sake of clarity, we are going to use long names for the comand-line switches. See the command-line help if you prefer short names for those cases in which a short name is available.
Bitmaps for the examples were generated as PNGs (Portable Network Graphics). If your browser is not ready for such format yet, you can visualize the PDF or PS versions by clicking on the links below each snapshot. Links to customization files, log files, GFF input files and output PostScript figures, are also available on each command-line shown.
Contents
|
Parsing BLAST Output
|
|
As we have seen in "Introduction to gff2aplot" tutorial, there are three basic formats in which we can provide alignment input data to the program. When visualizing data obtained with NCBI-BLAST (Altschul et al, 1997) or WU-BLAST (Gish, 1996-2003), we have to transform that output into GFF records. We can use parseblast, included within the gff2aplot distribution tarball. It can parse BLAST files into any of the three alignment formats described above.
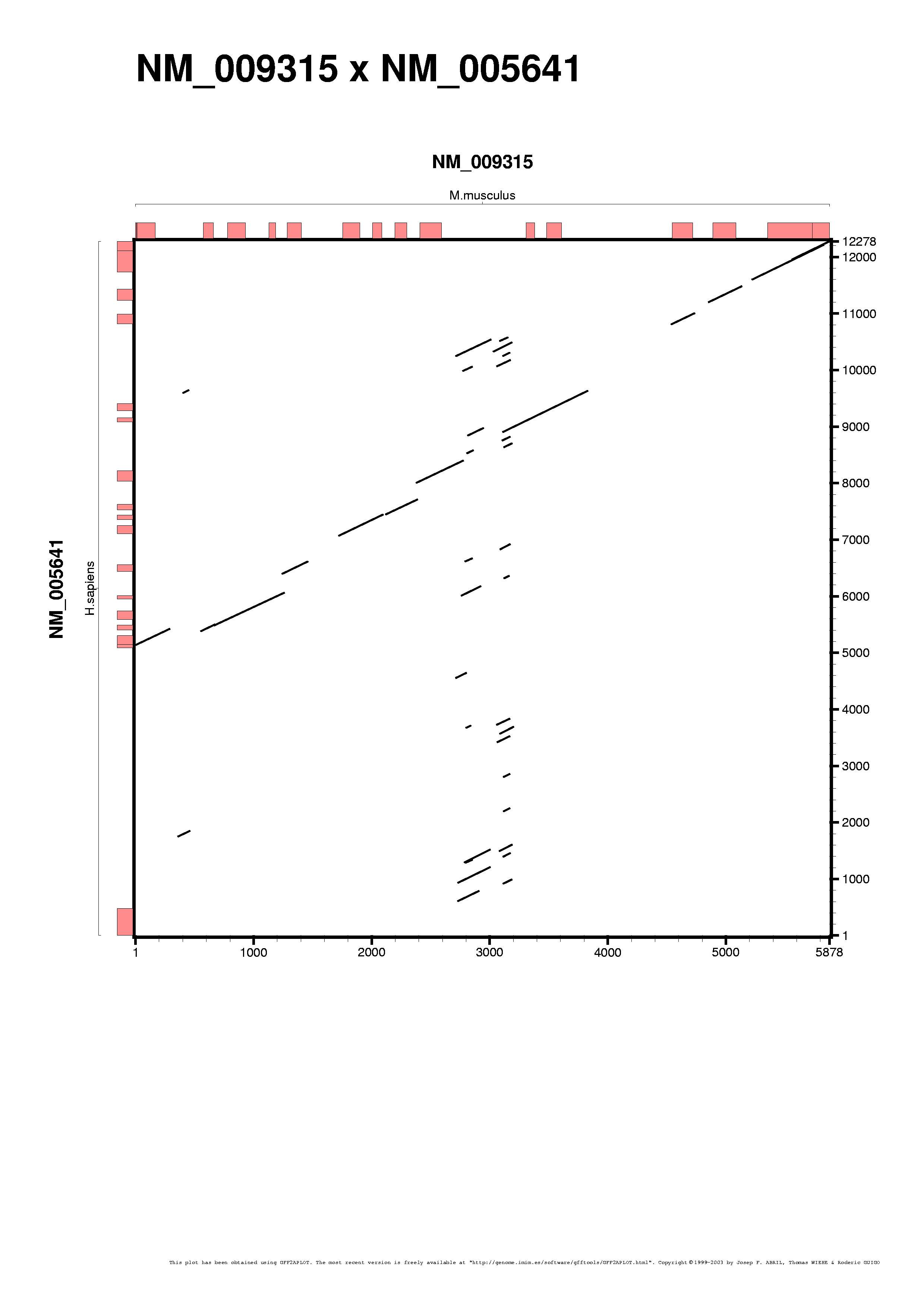
The "--aplot" command-line switch forces the output to APLOT pseudo-GFF format (see "E" case from the introductory tutorial). Input file was obtained using WU-BLAST BLASTN, TAF6 genomic region from human and maouse was compared (mouse NM_009315 as query and human NM_005641 as target sequences respectively):
Here, we process the same BLASTN input file, but we are going to produce GFF pseudo-version2 (see "D" case from the introductory tutorial). In that case a set of extra attibutes follows the grouping tag but do not conform to the tag-value model of attributes from GFF-version2. This can be achieved by simultaneously passing the "--fullgff" and "--compact-tags" command-line switches:
Still working with the same BLASTN file, if we want to get tight to GFF format version 2 (see "C" case from the introductory tutorial), to encode the alignment records, then "--fullgff" alone suffices, as shown below:
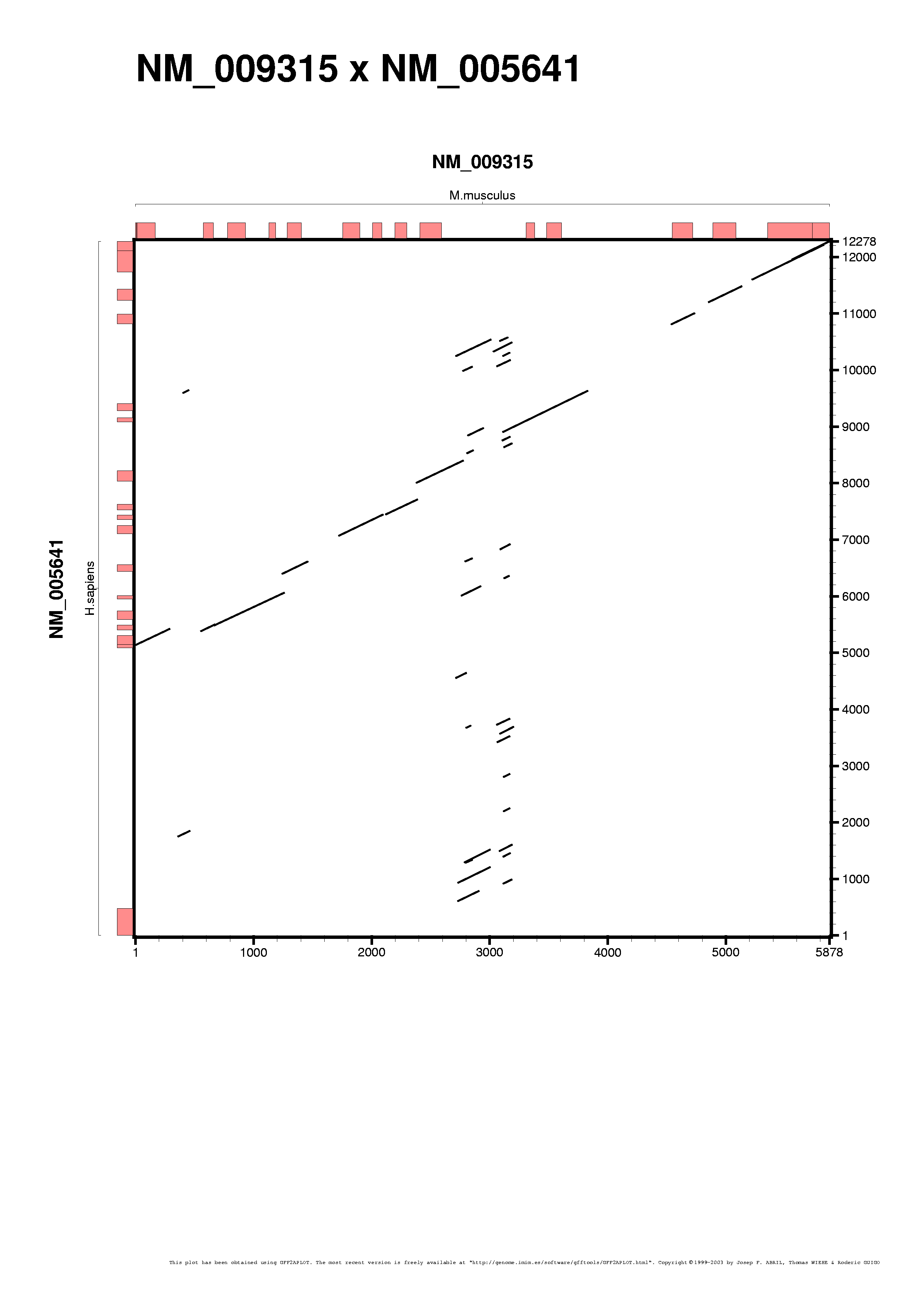
The following example takes input alignment from a WU-BLAST TBLASTX alignment (on the same sequences as for the BLASTN results), outputing APLOT records:
In all the examples, we have disabled to parse the frame from the BLAST input file. BLAST programs encode the frame as [ 1, 2, 3 ], but GFF standard requires it to be coded as [ 0, 1, 2 ]. parseblast is not able to recalculate the frame for reverse strand hits, as it will require the length of the corresponding sequence from the database (also known as targets). The output from BLAST tools only provides the length of the query sequence and the total length of the database. If you really need to obtain the frames for all the hits, you might require another program, not provided here, to recalculate them on the parseblast GFF output.
S. F. Altschul, T. Madden, A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D. Lipman
"Gapped BLAST and PSI-BLAST: a new generation of protein database search programs."
Nucleic Acids Research, 25(17):3389-3402, 1997.
W. Gish
WU-BLAST: http://blast.wustl.edu (1996-2003).
|
Raw Output
|
|
We run gff2aplot with the default settings on the WU-BLAST BLASTN results after being parsed with parseblast. The next two commands must produce the same plots, as the alignment data is the same (in compact GFF2 format and in APLOT pseudo-GFF respectively):
We do the same for the WU-BLAST TBLASTX alignment:
|
Modifying Plot Settings
|
|
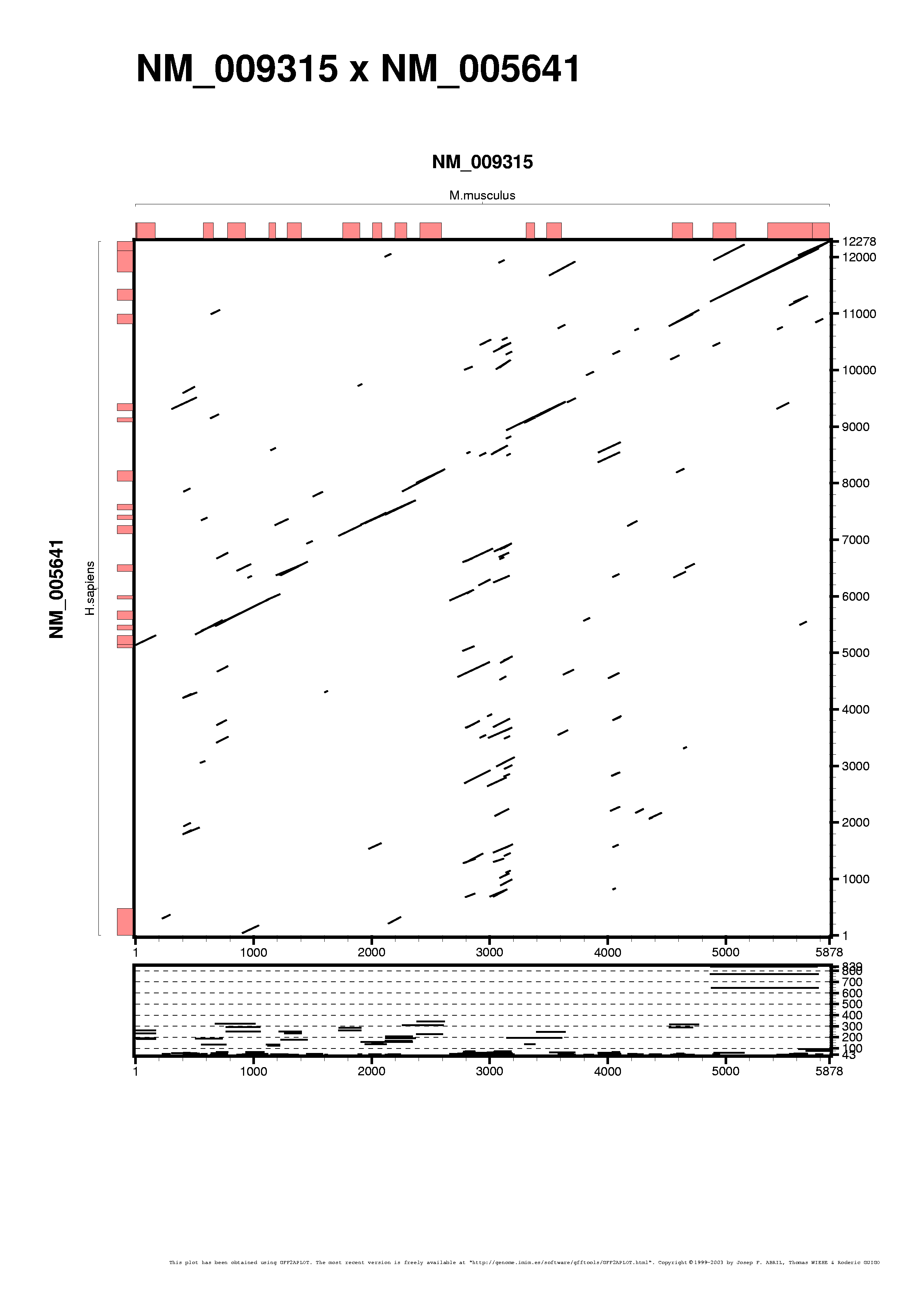
We are going to add some customization to the last plots, both the BLASTN and the TBLASTX one. We enable the axes annotation projections on the alignment panels. See the taf6.tblastx.rc customization file to see what we have changed.
|
Merging Data
|
|
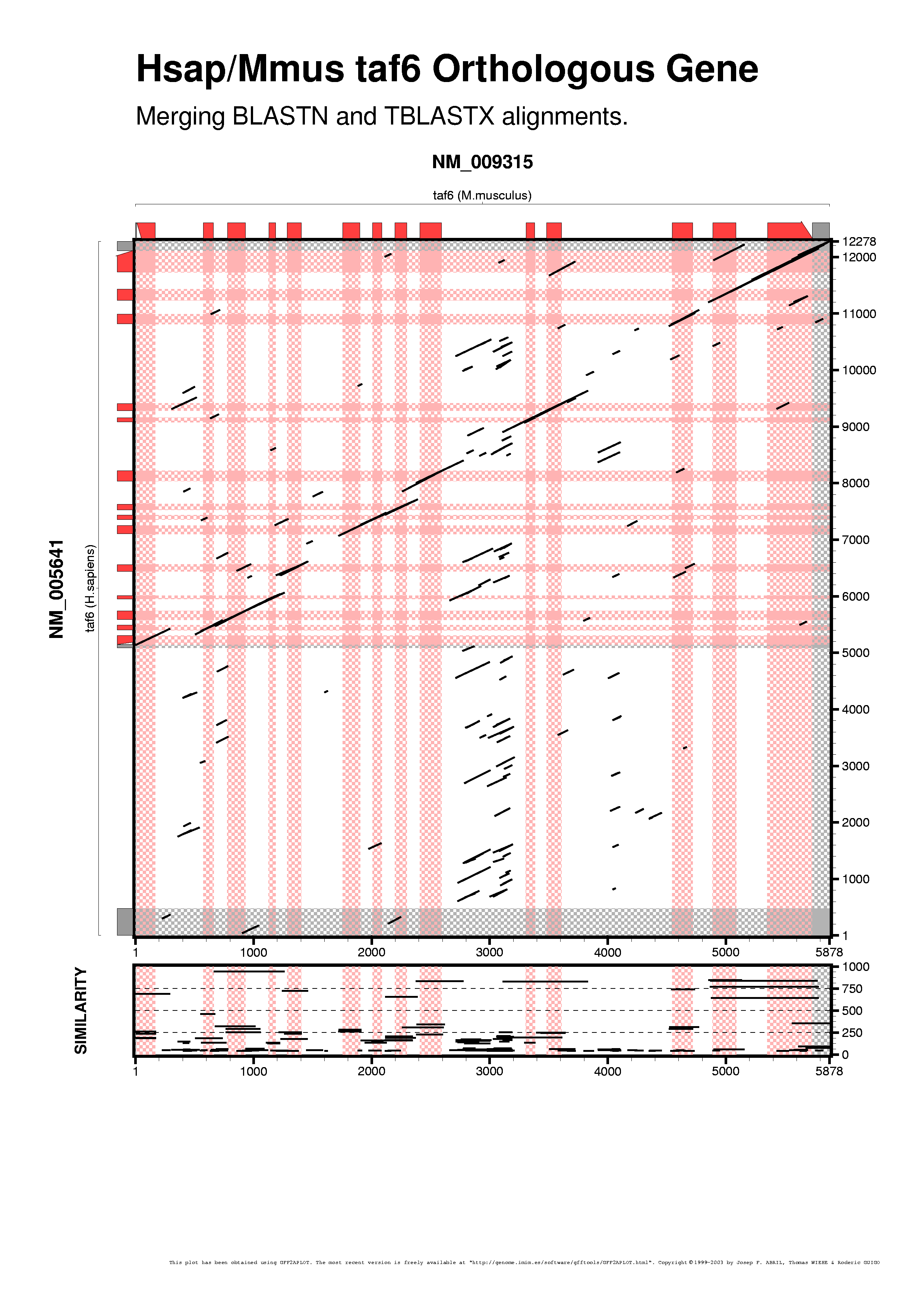
As gff2aplot does not depend on an underlying alignment algorithm, we can do things other tools cannot do, as merging results from different analyses. Here we are going to combine the BLATN and the TBLASTX alignments:
Showing all the alignment features with the same color does not provide us much information when merging datasets. We set different colors to distinguish between BLASTN and TBLASTX data, darkgreen and drakblue respectively, via command-line switches, as you can see in the next command-line:
|