| |
Summary
We will visualize the output produced by SIM on the genomic MHC syntenic region between human and mouse. We will use a filter to convert its output into GFF, the C program ali2gff is included in the gff2aplot distribution.
Once we have generated the whole genomic region figure, we will zoom into a single gene annotation. This will illustrate how you can expand a given area from your pictures, to highlight single features on multigenic annotation plots for instance.
NOTE.- For the sake of clarity, we are going to use long names for the comand-line switches. See the command-line help if you prefer short names for those cases in which a short name is available.
Bitmaps for the examples were generated as PNGs (Portable Network Graphics). If your browser is not ready for such format yet, you can visualize the PDF or PS versions by clicking on the links below each snapshot. Links to customization files, log files, GFF input files and output PostScript figures, are also available on each command-line shown.
Contents
|
Parsing SIM Output
|
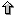
|
For illustrating this example we are going to visualize the alignment produced by the SIM program (Huang and Miller, 1991) on the syntenic MHCII genomic regions between human (hs.fa) and mouse (mm.fa). The sequences being analized contain four human genes (LMP2, TAP1, LMP7 and TAP2) and their counterpart ones in mouse (lmp2, tap1, lmp7 and tap2). The following is an excerpt from that program output, when applied to our data set:
| |
d {
"sim96 hs.fa mm.fa
M = 10, I = -10, V = -10, O = 60, E = 2"
}
s {
"hs.fa" 1 32000
"mm.fa" 1 35000
}
h {
">X87344"
">AF100956+AF027865"
}
a {
s 18340
b 863 804
e 19762 20115
l 863 804 890 831 79
···
l 19715 20068 19762 20115 85
}
···
a {
s 208
b 8426 33675
e 8541 33734
l 8426 33675 8449 33698 92
l 8506 33699 8541 33734 75
}
| |
Header fields contain information regarding program execution parameters, input fasta files provided and also the length of those sequences. Then you can find all the alignments and the locally aligned fragments that define each alignment.
That file can be translated into GFF records by using ali2gff, a C program which is also included in the gff2aplot distribution tarball. Here is an example of such a command-line:
Roughly speaking, "s" field defines the contents of the "seqbounds" GFF record. "s", "b" and "e" subfields within each alignment set ("a") define the contents of the "alignment" GFF records, while the "l" subfield corresponds to each "fragment" GFF records.
| |
X87344:AF100956+AF027865 sim96 seqbounds 1:1 32000:35000 . . .
X87344:AF100956+AF027865 sim96 alignment 863:804 19762:20115 18340.000 +:+ .:.
X87344:AF100956+AF027865 sim96 fragment 863:804 890:831 0.790 +:+ .:. ## cumulative nmp: 0.001
···
X87344:AF100956+AF027865 sim96 fragment 19715:20068 19762:20115 0.850 +:+ .:. ## cumulative nmp: 0.562
···
X87344:AF100956+AF027865 sim96 alignment 8426:33675 8541:33734 208.000 +:+ .:.
X87344:AF100956+AF027865 sim96 fragment 8426:33675 8449:33698 0.920 +:+ .:. ## cumulative nmp: 0.251
X87344:AF100956+AF027865 sim96 fragment 8506:33699 8541:33734 0.750 +:+ .:. ## cumulative nmp: 0.558
| |
On the other hand, we already have the axes annotation records from GenBank, we filtered them and saved into two GFF files, one for human (hs.gff) and one for mouse (mm.gff).
X. Huang and W. Miller
"A time-efficient, linear-space local similarity algorithm."
Adv. Appl. Math., 12:337-357, 1991.
|
Raw Output
|
|
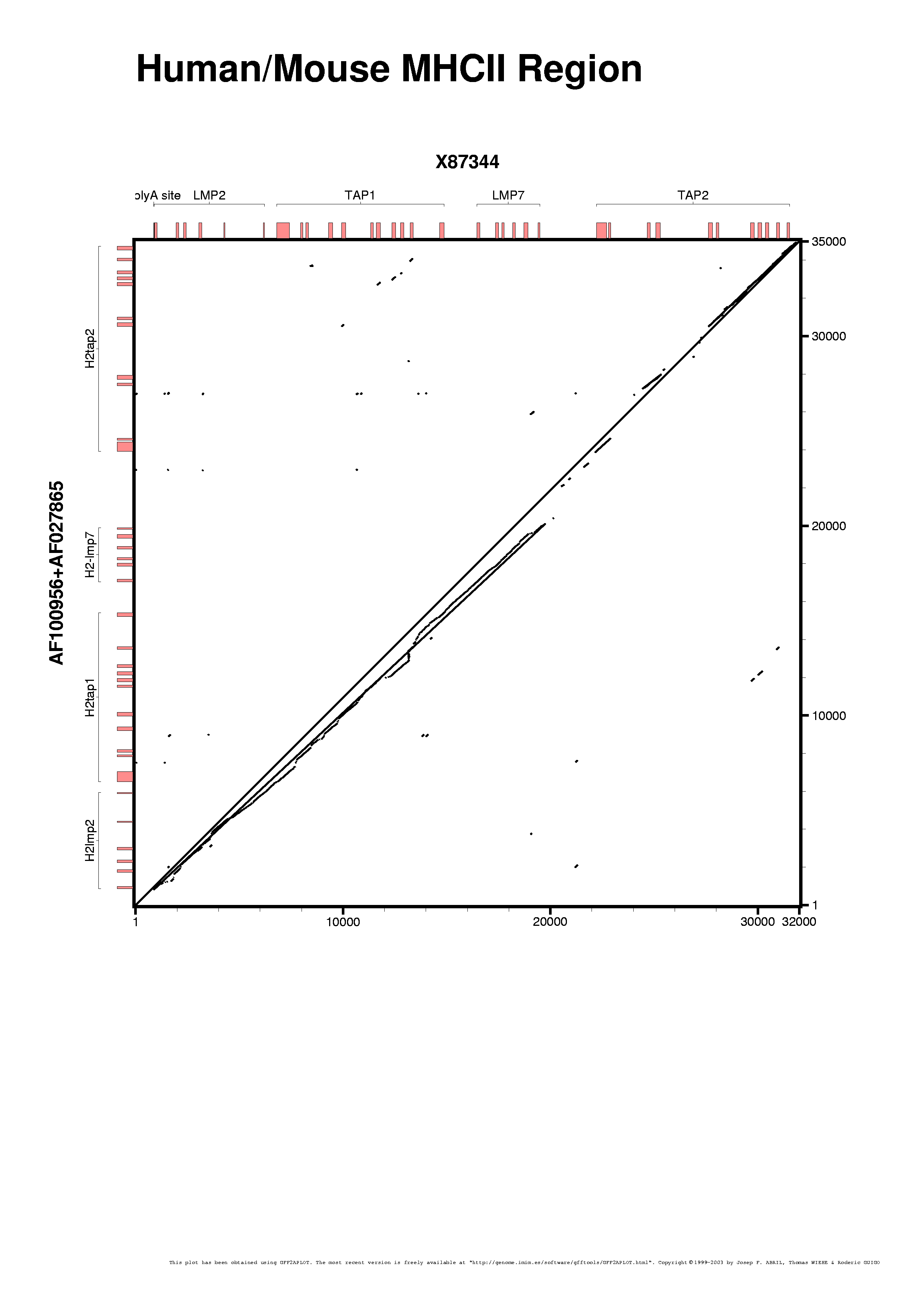
Putting together the human and mouse annotations with the alignment is an easy step. We are running gff2aplot with the default settings, just adding few labels to the figure via command-line switches ("--title", "--subtitle" and "--percent-box-label").
It is an extrange plot, isn't it ? We got an alignment diagonal which is an artifact product of the GFF input file we have used, although that file is well formated. It follows the APLOT pseudo-GFF format instead of the standard GFF one, but that must not alter the resulting plot.
Perhaps you already found what is wrong with that picture. Yes, we have included as an alignment the "seqbounds", the gff2aplot does not have a "fixed" definition of alignment records. The program takes all the records for two sequences that are in APLOT format as they code for alignment data to be drawn in the alignment panel. The same occurs with the GFF-version2 records having a pre-defined grouping tag, 'target "group-id"' (unless you have changed the alignment tag string via "align_tag" layout customization variable).
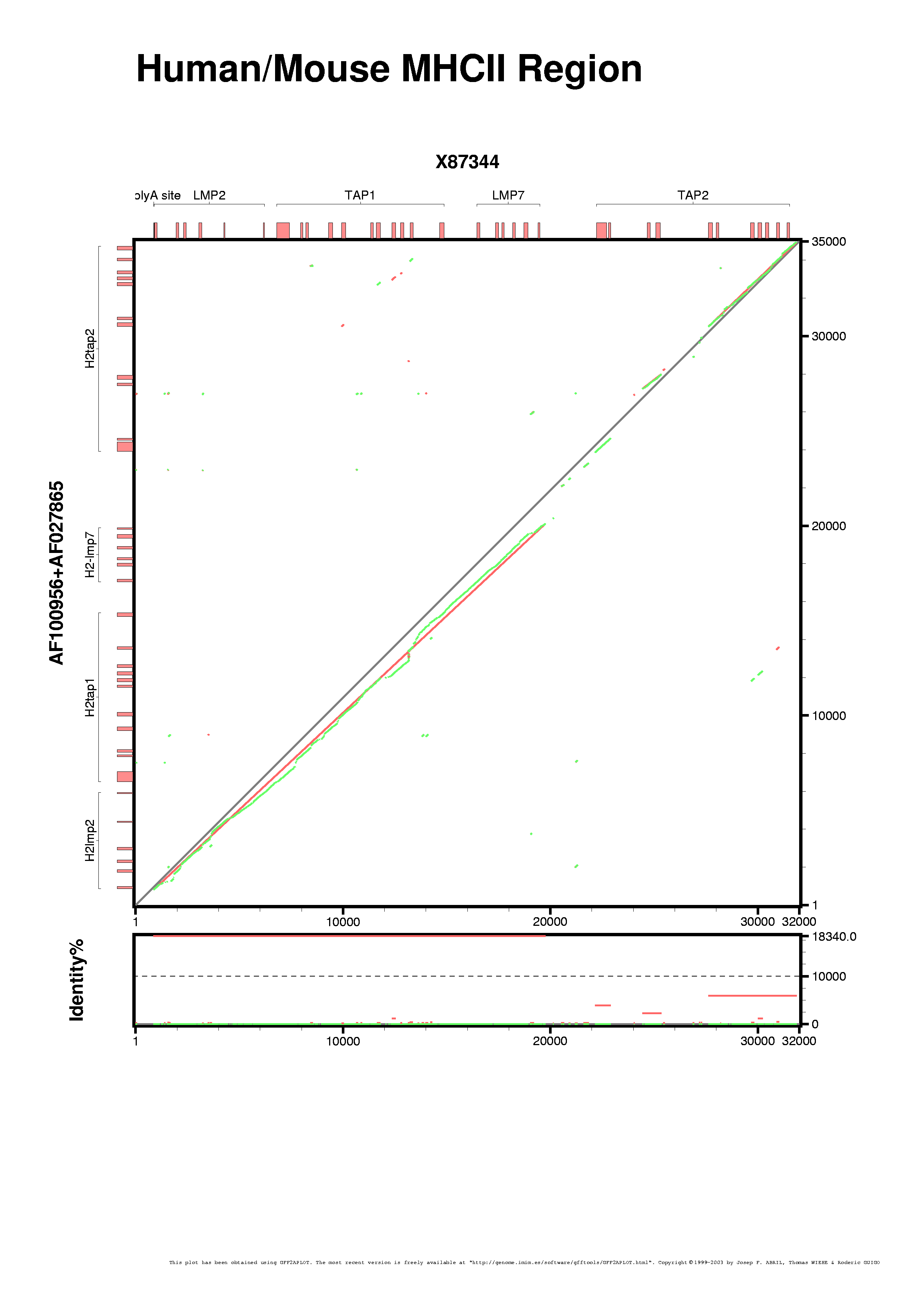
We are going to set different colors to the three GFF-features found in the "hs-mm.sim.gff" file ("seqbounds", "alignment" and "fragment"). We are also going to force that all those records without a defined strand, as "seqbounds" in this example, will be drawn in the bottomest layer. In the next plot we are setting both parameters from command-line (the "--feature-var" and "--strand-var" switches, respectively). We are switching on the percent bottom panel too (with "--show-percent-box").

[PNG] [PS] [PDF]
| |
gff2aplot.pl \
--verbose \
--percent-box-label "Identity%" \
--show-percent-box \
--title "Human/Mouse MHCII Region" \
--subtitle "" \
--feature-var "seqbounds::alignment_color=grey" \
--feature-var "alignment::alignment_color=red" \
--feature-var "fragment::alignment_color=green" \
--strand-var "..::strand_layer=0" \
--strand-var "++::strand_layer=2" \
-- hs.gff \
mm.gff \
hs-mm.sim.gff \
> hs-mm.sim2.ps \
2> hs-mm.sim2.log
| |
|
Finishing Plot for MHC Region
|
|
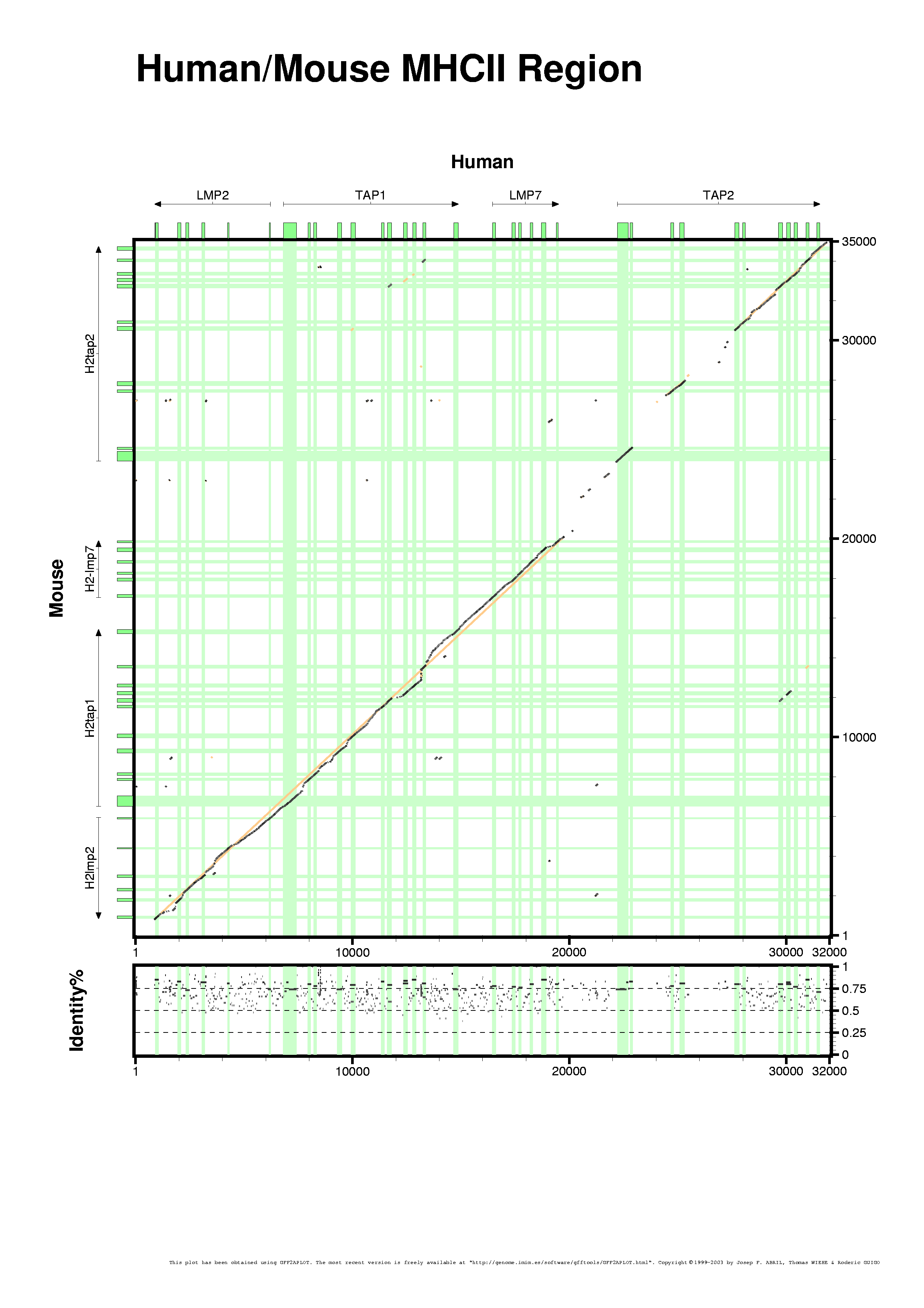
To make the last figure we have changed few customization options to highlight an artifact, the "seqbounds" GFF record on the input file. We can remove those fields that are not related with the alignment after generating the GFF file, or we can hide those features that are not relevant for the final picture. We do not want to show the "seqbounds" records although they may be useful for other programs using the same GFF file, maybe in such cases we do not want to remove those records from the file. There is an alternative solution, just hide all the records containing such features. The following picture only shows the alignment fragments. We have defined that "seqbounds" and "alignment" features must be hidden, in this case by setting the correpsonding variable in the customization file (see mhc2.rc).
We have said that we were going to hide the "alignment" GFF-features from the plot and we just changed its color. What's happening here ?
We simply provided an extra customization file (see mhc2spc.rc). That file contains two variable settings which override the ones defined in the previous customization file (mhc2.rc). It is a must to notice here that the order of the customization files (as for the GFF input records) is very important. We do not get the same results if we swap the command-line switches and we provide "mhc2spc.rc" before "mhc2.rc". The example also illustrates the advantatges of having a main customization file upon which we can change paramters of the figure (mainly via command-line switches, but via extra customization files too).
|
Zooming Into the LMP2 Gene Region
|
|
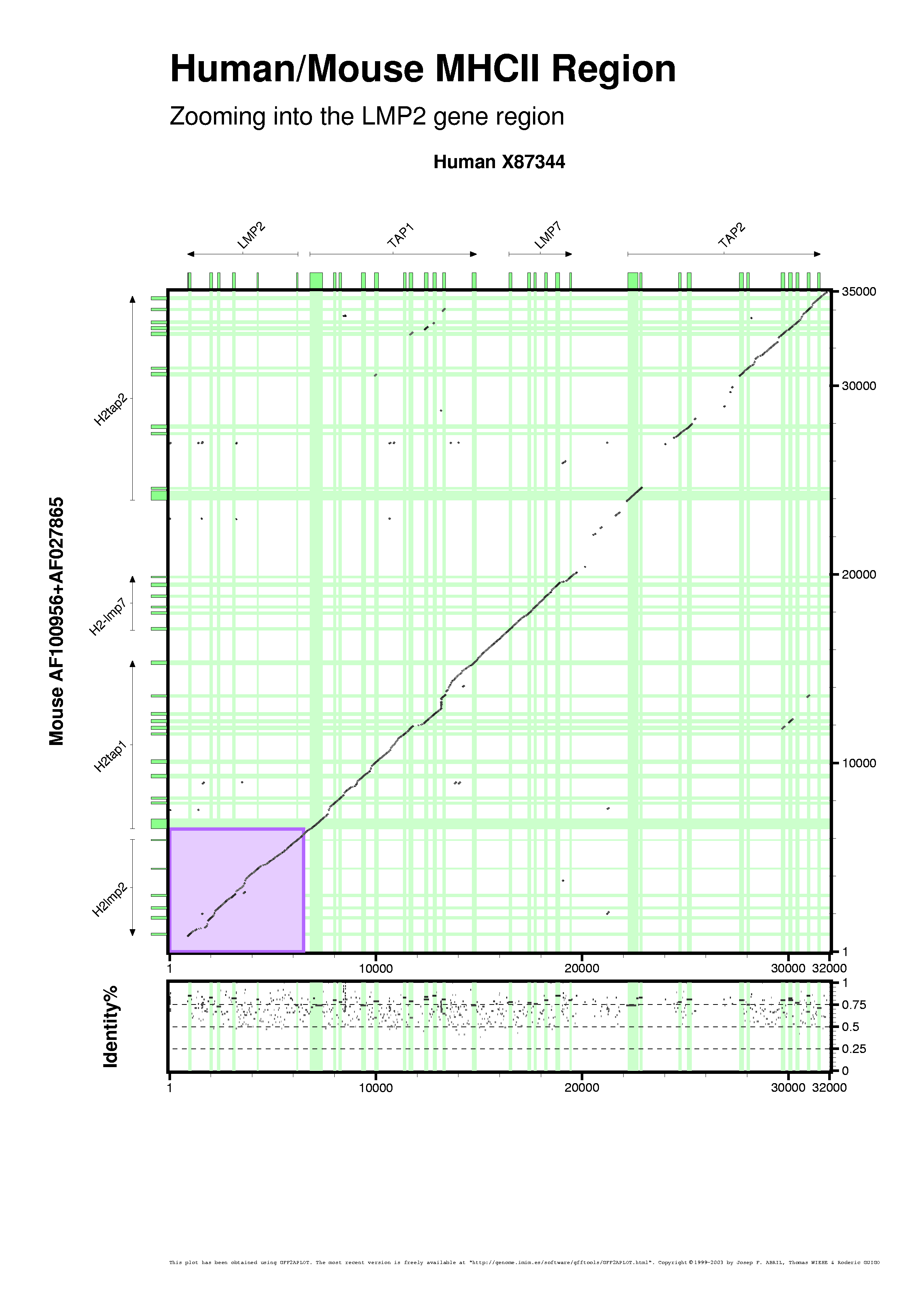
In this tutorial we would like to introduce the "zoom" feature too. When working with large datasets we can take two approaches: making larger plots (I mean using larger paper sizes) or zooming into interesting plot regions. There are two command-line switches that make that option possible without modifying the GFF input records (for instance, by filtering those records outside the zoomed region boundaries). "--zoom-area" allows user to show where is the area being expanded and "--zoom" which performs the real zoom into that area. Both can take up to four optional parameters, set from command-line or customization files, that define the X and Y nucleotide coords of the enlarged plot region. In this case, we are assuming that the zoom area will start on both sequences origins, otherwise you can define that with "--start-x-sequence" and "--start-y-sequence" command-line options. "--end-x-sequence" and "--end-y-sequence" define the final coords for the zoomed area.

[PNG] [PS] [PDF]
| |
gff2aplot.pl \
--verbose \
--percent-box-label "Identity%" \
--show-percent-box \
--title "Human/Mouse MHCII Region" \
--subtitle "Zooming into the LMP2 gene region" \
--zoom-area \
--end-x-sequence 6500 \
--end-y-sequence 6500 \
--x-label "Human X87344" \
--y-label "Mouse AF100956+AF027865" \
--custom-filename mhc2.rc \
--custom-filename mhc2labels.rc \
-- hs.gff \
mm.gff \
hs-mm.sim.gff \
> hs-mm.sim.zbox.ps \
2> hs-mm.sim.zbox.log
| |
Above picture highlights the area being enlarged in the plot below. We defined the fill color of the highlighted area (set to violet in mhc2.rc) to be the same for the bottom figure (set to violet in mhc2+sgp1.rc). We are including gene predictions obtained by SGP1 program (Wiehe et al, 2001). In both plots, the group (say here "genes") labels angle is set on "mhc2labels.rc" customization file.
T. Wiehe, S. Gebauer-Jung, T. Mitchell-Olds and R. Guigó
"SGP-1: prediction and validation of homologous genes based on sequence alignments."
Genome Research, 11(9):1574-1583, 2001.
|
|