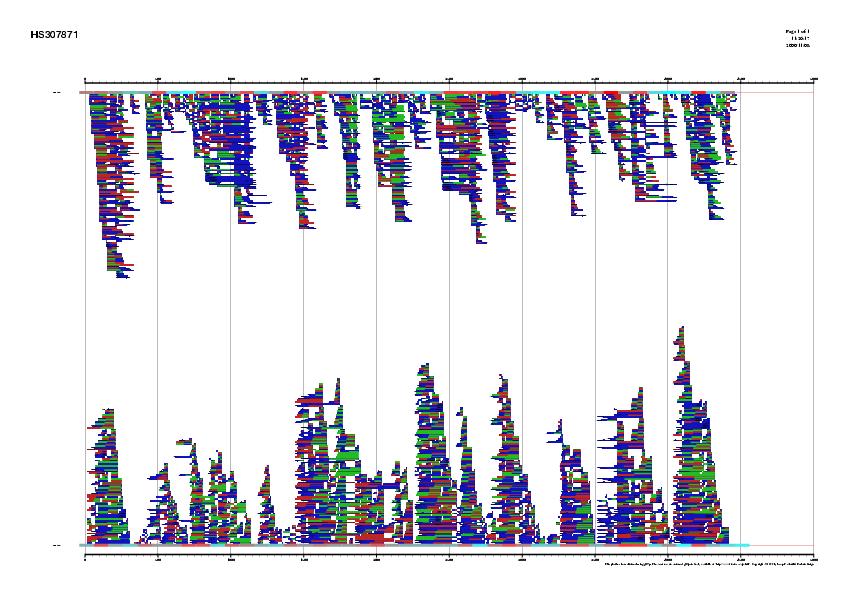
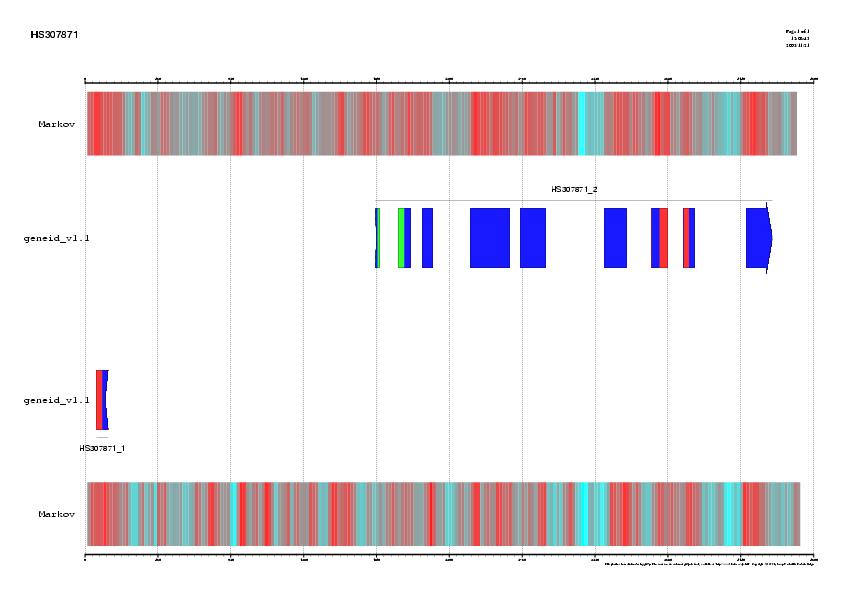
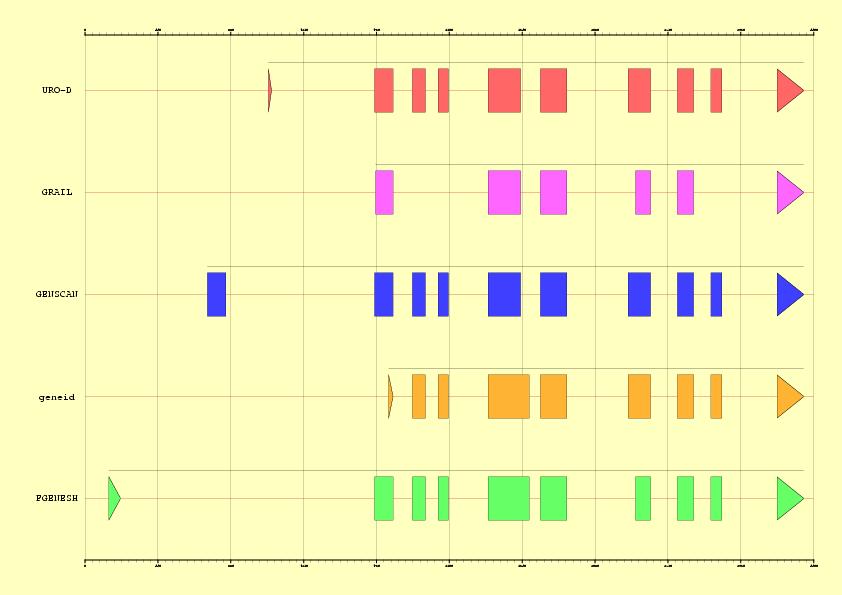
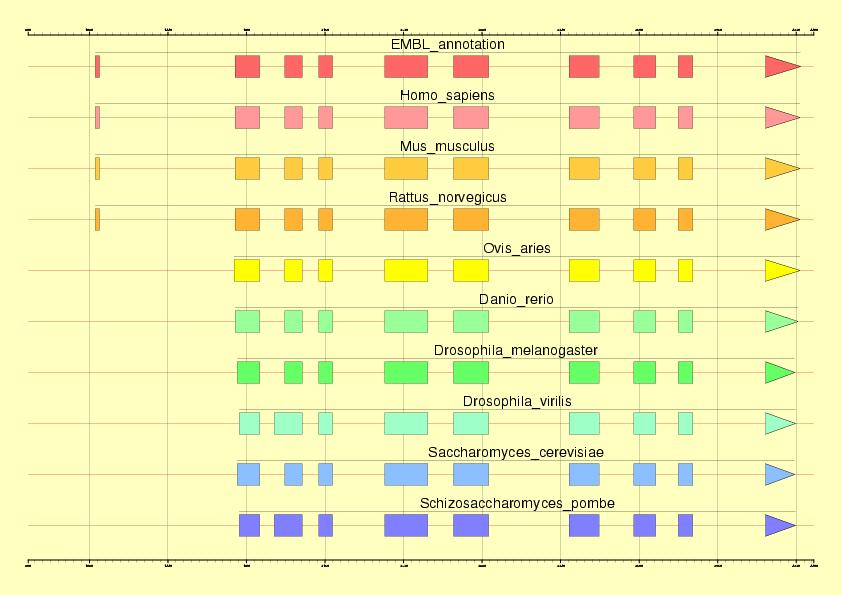
Abstract: In this exercise, a previously annotated gene will be used to
measure the accuracy of different gene finding approaches. GRAIL,
GENSCAN, geneid, FGENESH, GenomeScan, GrailEXP and GENEWISE
will be used to annotate the sequence. Both search by signal, content and
homology (protein and cDNA sequences) methods will be employed in order to
improve the ab initio results. Weak conservation of Start codons will lead
to wrong prediction of initial exons in most cases.
Colour legend:
Genomic element
Operations or links
|
Step 4. Using GrailEXP
- Connect to GrailExp homepage
- Activate Galahad EST/mRNA/cDNA Alignments box
- Select GrailEXP database (RefSeq/HTDB/dbEST/EGAD/Riken)
- Activate exon assembly: Gawain Gene Models
- Paste DNA sequence
- Press Go!
button
Check the results: predictions and supporting information
Compare annotations, ab initio GRAIL prediction and five predicted
alternative spliced variants

|
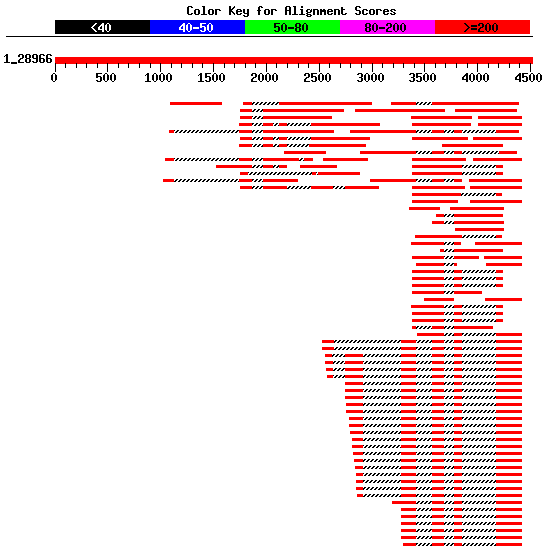
Figure 3. Comparison between EMBL annotation and genes predicted ab inition by Grail Vs five alternative predictions supported by ESTs information
in the sequence HS307871
|
Step 5. Using other gene finding programs + alignment of transcripts
Using blastn, we can search
the database est_human for ESTs supporting
future predictions. Filter this output in order to select those
non-overlapping ESTs that could form a complete cDNA sequence (see Figure 4).
Moreover, ESTs not divided into two or more pieces in the genomic sequence
(containing a couple of splice sites) should be rejected.
- Connect to the FGENESH-C server
(on Gene finding with similarity menu)
- Paste the sequence HS307871
- Paste the cDNA sequence or EST you have selected
- Press the search button
- Notice that predicted gene will necessarily supported by homology
information, so it will likely mapped only in the genomic region overlapping
your EST query.
 |
|
Figure 4. Best human ESTs in the alignment mapped on the genomic sequence HS307871
|
|
|
D. Using protein homology information
|
Step 6. Spliced alignment
Spliced alignment is very useful when we have additional information
(a putative homologous protein sequence) about the content of the sequence.
Thus, gene prediction is guided by fitting the protein sequence into the
best splice sites predicted in the genomic sequence.
- Open the NCBI blast server
- Choose blastx program (genomic query versus protein database)
- Paste the genomic sequence and press the
Blast! and
Format!
- Select the first protein. Display the FASTA sequence or click
here. Obviously, it is the real
protein annotated in the genomic sequence.
- Open genewise web server
to use this protein to predict the best gene structure
- Paste both protein and genomic sequences and run the program
- Compare predicted gene (end of the file) and annotations: look
for splice sites within introns to check exon boundaries are correct
 |
Figure 5. Best HSPs representing proteins homologues similar to the
genomic sequence HS307871 obtained using blastx
|
|
Step 7. Spliced alignment using homologous proteins
From blastx output, choose several homologous genes and run genewise for each
one separately, again. Observe the gain of accuracy as long as the homologue is closer to the original human protein:
|
Step 8. Using protein homology information: GenomeScan
Protein homology information can also be used to enhance ab initio predicted
exons supported by blastx HSPs as in the case of GenomeScan and
geneid improving therefore the final prediction
GenomeScan:
- Connect to the
GenomeScan web server
- Retrieve the protein from the previous blast search
- Paste both genomic and protein sequences
- Press the button GenomeScan
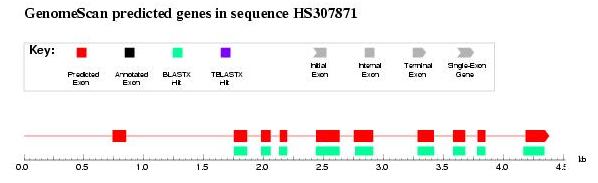
- Check the results. It seems that the first exon has not been detected
even using homology information. This is due to the fact that blast programs
have a minimal word lenght.
 |
|
Figure 6. GenomeScan output: first exon is not correctly predicted probably due to blast length restrictions
|
|
|
|
E. Using a genome annotation browser
|
Step 9. Golden path archive:
- Open the
UCSC Genome Bioinformatics Site
- Select the blat link to locate
the genomic coordinates of our sequence
- Paste the DNA sequence in FASTA
format (HS307871)
- Submit the file
- Click over the first hit: (browser link)
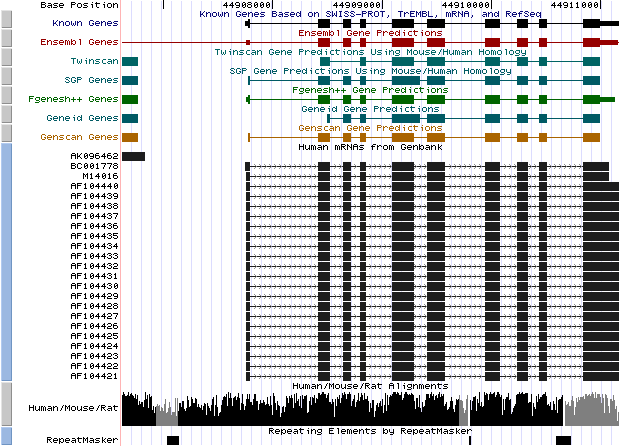
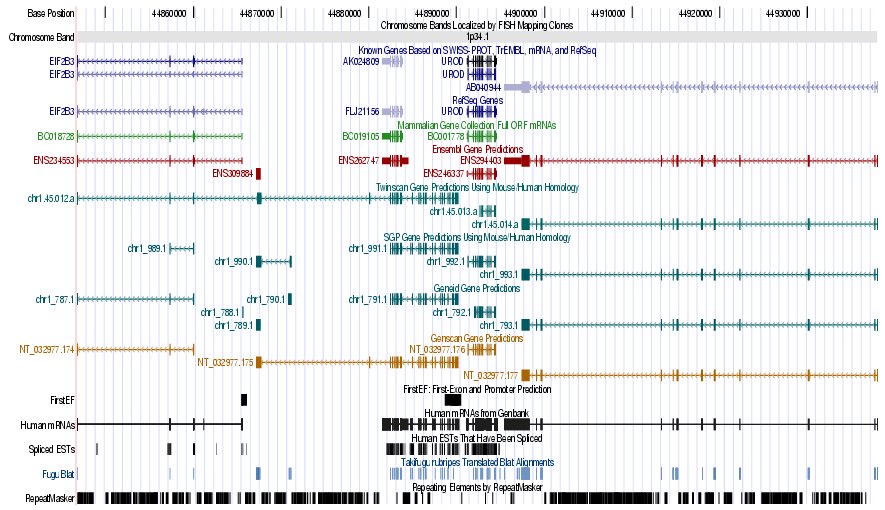
- Compare the graphical annotation with the EMBL entry of the gene
- Analyze these different sets of output options:
Genes and Gene Prediction Tracks,
mRNA and EST Tracks
 |
 |
|
Figure 7. (a) UCSC genome browser representation of the region
containing the gene uroporphyrinogen decarboxylase (URO-D) (b)
UCSC genome browser representation of the contex (100Kbps) region around
the gene uroporphyrinogen decarboxylase (URO-D).
|
|
|
|
F. Results
|
Here you can find the solutions to every exercise:
|
|
|
F. Bibliography
|
- J.F. Abril and R. Guigó.
gff2ps: visualizing genomic annotations. Bioinformatics 16:743-744 (2000).
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 215:403-410 (1990).
- Burge, C. and Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78-94 (1997).
- E. Blanco, G. Parra and R. Guigó.
Using geneid to Identify Genes. In A. D. Baxevanis and D. B. Davison,
chief editors: Current Protocols in Bioinformatics. Volume 1, Unit 4.3.
John Wiley & Sons Inc., New York. ISBN: 0-471-25093-7 (2002).
- G. Parra, E. Blanco, and R. Guigó.
Geneid in Drosophila. Genome Research 10:511-515 (2000).
- Asaf A. Salamov and Victor V. Solovyev. Ab initio Gene Finding in
Drosophila Genomic DNA Genome Res. 10: 516-522 (2000).
- Yeh, R.-F., Lim, L. P. and Burge, C. B. Computational inference of
homologous gene structures in the human genome. Genome Res. 11: 803-816 (2001).
-
D. Hyatt, J. Snoddy, D. Schmoyer, G. Chen, K. Fischer, M. Parang, I. Vokler, S. Petrov, P. Locascio, V. Olman, Miriam Land, M. Shah, and E. Uberbacher.
Improved Analysis and Annotation Tools for Whole-Genome Computational Annotation and Analysis: GRAIL-EXP Genome Analysis Toolkit and Related Analysis Tools.
Genome Sequencing & Biology Meeting (2000).
- Ewan Birney and Richard Durbin. Using GeneWise in the Drosophila
Annotation Experiment. Genome Res. 10: 547-548 (2000).
|
|