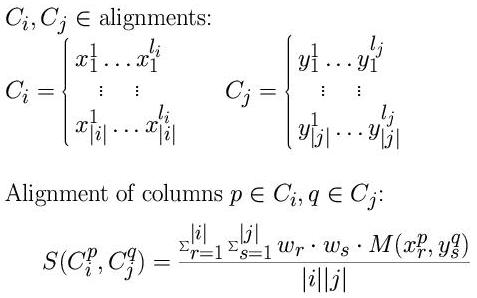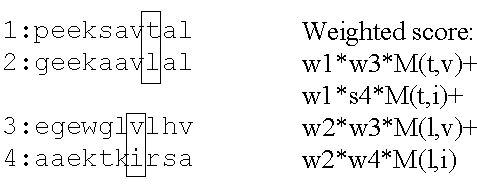Features:
- Progressive alignment (neighbour-joining method)
- Assign weights to the sequences to correct unequal sampling
across all evolutionary distances in the data set
- Different substitution matrices on every stage of the alignment
- Position-specific gap penalties
- Addition of new sequences to an existent msa
- Delay the incorporation of divergent sequences
1. Construction of the distance matrix:
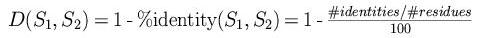
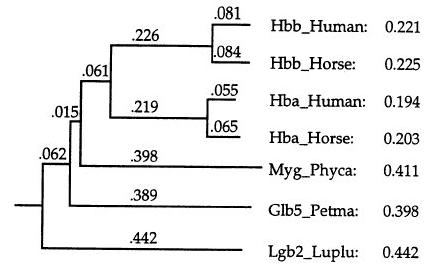
2. Construction of the guide tree:
- Neighbour-joining method: unrooted tree
- Mid-point method: place the root at a position where the
means of the branch lengths on either side of the root are equal
- Derive a weight for each sequence (up/down) to avoid
duplicated information
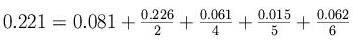
3. Alignment:
|
The score between a position of one alignment and one from
another is the average of all the pairwise substitution matrix scores from
the residues in the two sets of sequences multiplied by the weight of
the sequences.
|
- Gap penalties (opening and extension) -
GOP: (the substitution matrix, the similarity and the length of sequences)
GEP: (the difference in length of the sequences)
Position specific GOP: existing gaps, near existing gaps,
hydrophilic residues
- Substitution matrices -
Choice of different PAM/BLOSUM according to the distance of the sequence
or groups of an alignment

|
|