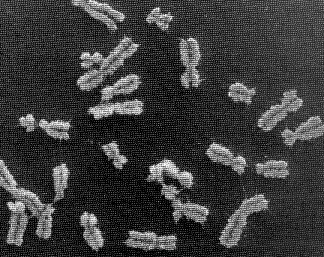
Human chromosomes
In what follows, we revise the key concepts regarding the eukaryotic gene and genome structure needed to understand the annotation of genomes. In this context, annotation refers to the description and location of genes and other biologically relevant features of a genomic sequence. Our main goal is to fully comprehend what genome annotation projects offer and, just as important, what they do not yet provide.
In this regard, it's worth noting that current gene prediction programs, among other bioinformatics tools, systematically ignore the complexity of eukaryotic gene structure. Diversity comes from alternatively spliced genes, non-canonical signals (that affect either splicing or translation) and from regions that control gene transcription, promoters, which are not yet well understood. Here, we give and overview to see some of the limitations and future directions in the gene prediction field.
The genome is the genetic material of an organism, that is, the total
amount of DNA in the cell. In eukaryotes, it is usually organized into
a set of chromosomes, which are extremely long chains of DNA that are highly condensed. In the picture below, human DNA is shown packaged into chromosome units (as seen during mitotic metaphase). Note the sister chromatids (that contain identical daughter DNA molecules), centromeres and telomeres.
It is time to introduce the three main genome browsers and check which genomes they are serving. In this practical, we will stick to the Ensembl server, but feel free to browse the others later on.
Questions:
Now, select human and make sure you understand the main concepts in this page. Contrary to the view reported in newspapers, the genome sequence and its corresponding annotations are highly dynamic. There are two levels at which data can be updated: 1) sequence level and 2) annotation level.
Questions:
DNA molecules consist of two anti-parallel chains held together by complementary base pairs that form a double helix. This structure is of major importance for computational analysis and, to a great extend, determines how it can be performed:
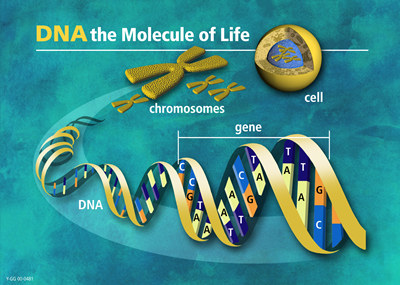
Note that, in this course, we only analyze DNA at the sequence level. So, let's now check it. Please, select the chromosome 21 in the karyotype plot.
Questions:
Now, let's search for a specific gene over the whole genome. In the Find window, lookup the alcohol dehydrogenase gene.
Questions:
However, before analyzing the alcohol dehydrogenase gene, we will take a look at the eukaryotic gene structure, processing and expression (see below).
Transcription, splicing and translation are the main processes that account for the expression of protein coding genes. Each step is directed by sequential and structural signals. In what follows, we describe from both the biological and computational point of view, how these sequence motifs are used to go from DNA to RNA to the final protein product.
The schema below, highlights these processing steps:
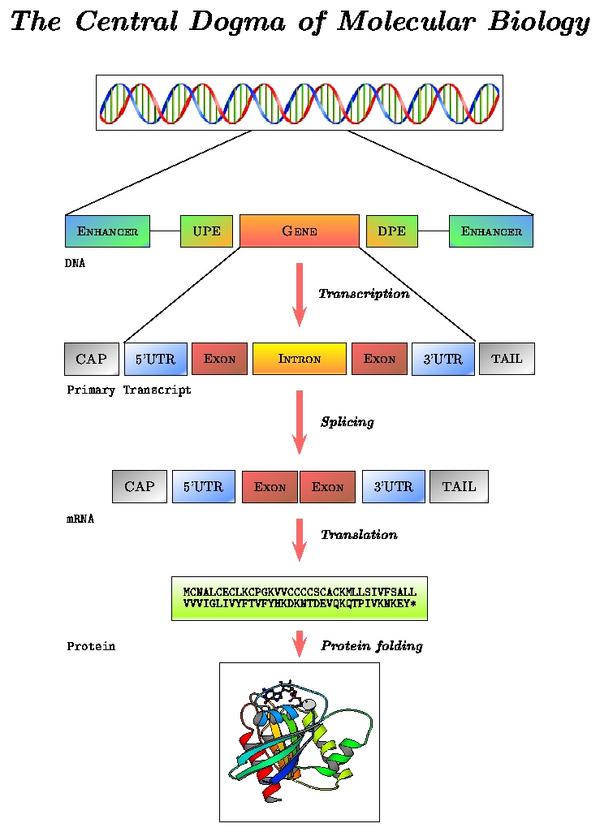
Locate the "Transcript Structure" section in the alcohol dehydrogenase gene report. Note the correspondence between levels of reported data and processing steps:
We will follow these links in the order shown, but first, move to the text below for a more precise discussion of the eukaryotic gene structure.
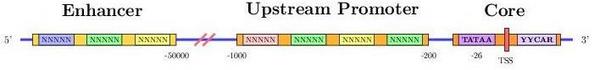
Transcription starts when a region upstream of the gene (promoter
region) is activated (bound) by transcription factors. These regions,
control whether a gene is transcribed from the forward or reverse
strand. In any case, the strand which is actually transcribed is
called template or sense strand and the other, nonsense or antisense
strand.
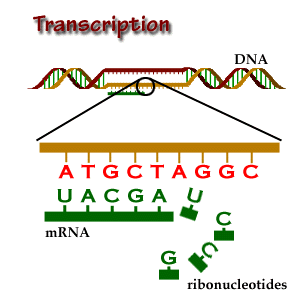
In short, transcription is the copying of DNA (template strand) to RNA
(pre-mRNA). However, when analyzing mRNA, cDNA or EST data, bear in
mind that the mRNA to be translated is, in sequence, identical to the
coding strand (coding here always refers to translation, and not to
transcription). That is, the mRNA is transcribed from the strand that
has its complementary sequence. In conclusion, when annotating
genomes, genes are annotated in relation to their coding
strand.
There are three main types of transcript data:
The transcription process brings us to two relevant terms in relation
to the study of gene regulation: cis and trans
elements. A locus is a cis-acting element if it must be on the same
DNA molecule in order to have its effect. Transcription factor binding
sites are a good example of cis-acting regulatory elements. A locus is trans-acting if
it can effect a second locus even when on a different DNA
molecule. Transcription factors are a good example of trans-acting
regulatory elements. Note, again, that these terms are distinct from the
notions of upstream and downstream region explained above.
Another biological example is the so-called trans-splicing, where
exons from different transcripts are spliced and joined together. That
is, elements from independent sequences end up acting together in the
same mature mRNA.
Eukaryotic genes are short DNA stretches within a genome with a
peculiar and discrete structure.
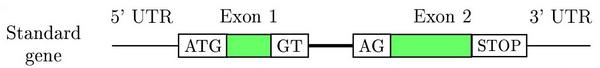
Gene prediction programs make use of this structure to find genes in a genome. The main characteristics are:
Follow the Exon information link.
Questions:
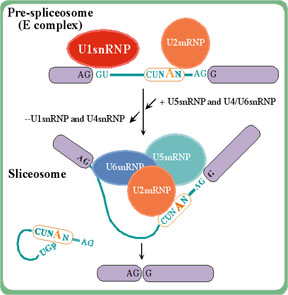
Splicing is an RNA-processing step in which introns in the primary
transcript are removed. Splicing signals, GT (donor) and AG (acceptor)
in the intron region, are used to delimit exon-intron boundaries, so
that exons (coding and non-coding ones) are joined together. In this
way, the open reading frame sequence along with the 5' and 3'
Untranslated Regions (UTRs) are ready to be processed by the
ribosome.
Follow the Transcript information link.
Questions:
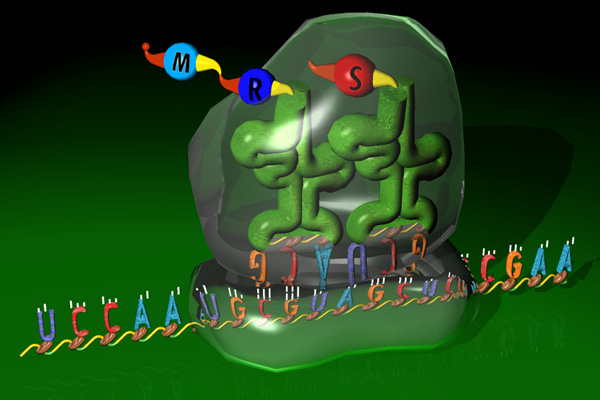
In translation the mature mRNA sequence is translated into a
protein. Again, the ribosomal machinery is guided by several signals
along the mRNA sequence to find the right open reading frame (ORF) and
to determine where the translation should terminate.
Follow the Protein information link.
Questions:
Let's now try to get a deeper insight into the biological role of this gene. We will connect to a couple of web-based resources to learn more about our gene and protein. GeneCards is a database of human genes, their products and their involvement in diseases. Connect to this database and search GeneCards by symbol using the HUGO id corresponding to the adh gene: AKR1A1 (as shown in the Ensembl gene report).
Questions:
Finally, we will try to get other possible identifiers for this gene in several databases. Connect to GeneLynx and do a quick search in human for the HUGO ID: AKR1A1.
Questions:
It is about time to fully use the main feature of genome browsers:
the ability to display all available information along a genomic
region of interest. However, first take a look at the picture below to
make sure you understand the pipeline behind these data.
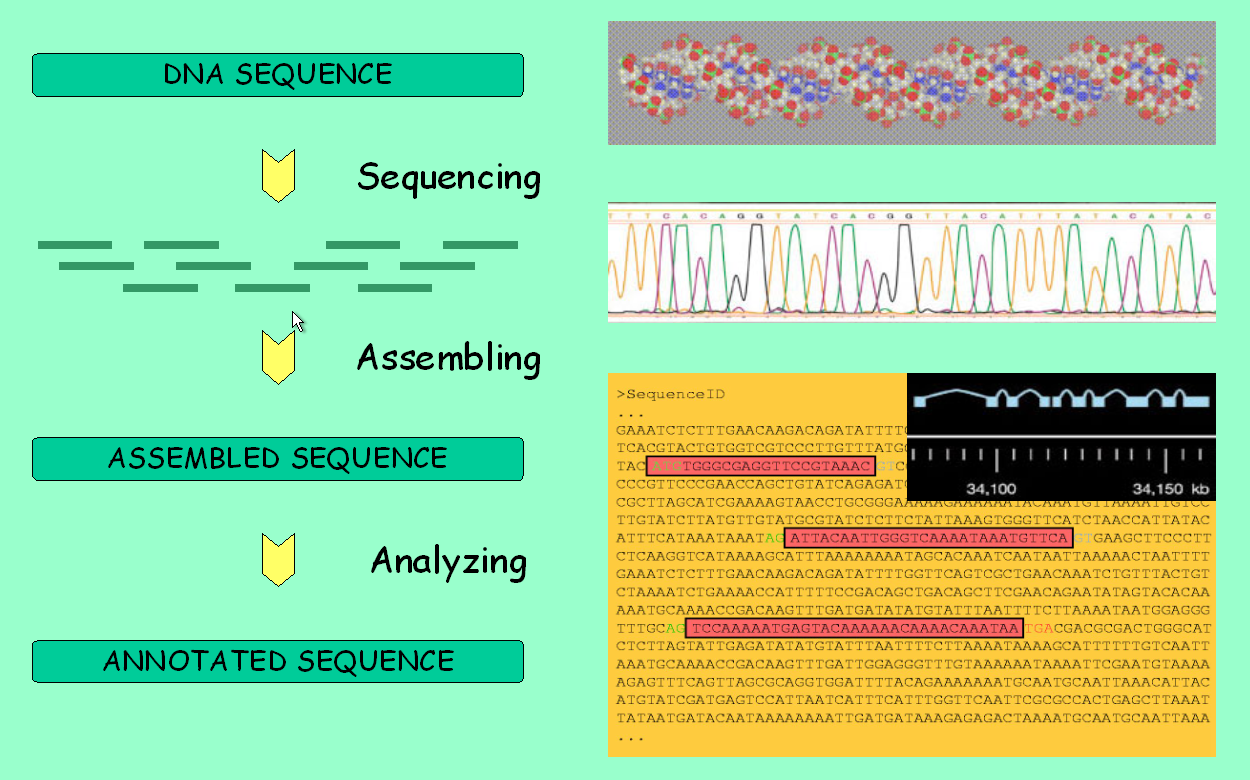
We will browse the genomic region of the adh gene that we are working with. From the Gene Report page, follow the link with the genomic location. There are four levels of resolution:
The mouse and rat synteny tracks above call for a brief discussion of this concept. Although, historically, synteny means "in the same strand" and syntenic genes are those in such a disposition, but syntenic regions between genomes are understood as regions in which gene order is conserved and, therefore, syntenic genes are putative homologues that have an orthologous, paralogous or even xenologous relation. See below for a short definition of these key but often misinterpreted concepts.
Go to Chromoview in the Ensembl web-site by clicking on a chromosome. From here we can link to Synteny view, which offers a chromosomal view of the synteny between genomes.
Homologous genes are genes that are related through a common
evolutionary ancestor. Homology is usually inferred on the basis of
sequence similarity but bear in mind that, through random and
convergent evolutionary processes, biological sequences can share a
reasonable degree of similarity without a true evolutionary
relationship. In addition, it is incorrect to say that a pair of
related genes are, for example, 80% homologous, because genes are
either evolutionarily related or not. On the other hand, one can speak
of a percentage of sequence similarity between genes.
This is of importance, for instance, when reading a blast output and deriving evolutionary implications. The score and the e-value are based on the similarity between sequences. However, this does not necessarily ensure a close phylogenetic relationship, although it suggests one.
Orthology, paralogy and xenology are homology subtypes. That is, they define a specific type of relationship between genes over space and time. Read these definitions carefully:
This paper discusses these concepts in more detail.
The transcription start site, can vary in the same gene depending on how the promoter region is activated. This results in a different pre-mRNA and, potentially, a differentially expressed mRNA or even a distinct protein may be achieved.
Briefly, alternative splicing is an important cellular mechanism
that leads to temporal and tissue specific expression of unique mRNA
products. This is accomplished by the usage of alternative splice
sites that result in the differential inclusion of RNA sequences
(exons) in the mature mRNA.
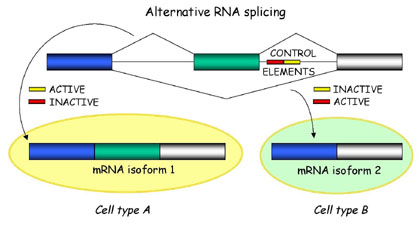
In general, current gene prediction programs cannot predict alternative mRNAs in a reliable way, unless transcriptional data (mRNAs, cDNAs and ESTs) are available.
Translation by the ribosome is a complex process. Sources of variability are:
Try to complete this practical.
