NOTE: click on the images through this document to download higher quality postscript images.
An issue that naturally arises is that of the reliability of the predictions obtained by Gene Identification and Gene Structure prediction programs. The issue concerns both users and developers. It is particularly important for the users confronted with the need to identify those functional domains potentially encoded in large uncharacterized genomic regions. Indeed, experiments are often planned on the basis of such predictions, and these may involve a considerable amount of effort and resources. It is also important for the developers in order to assess the actual status of the problem. Moreover, as we are entering the large-scale sequencing phase of the Genome Projects, with an expected rate of production of over two megabases daily during the next few years, annotation of genomic sequences will need to be partially automated. Automatic annotation, however, is only possible if highly reliable gene prediciton programs exist.
Burset and Guigó (Genomics 34, 353-367, 1996) attempted to develop data set and performance metrics standards for consistently testing of Gene Identification software. They tested a number of programs on a large set of vertebrate sequences with known gene structure, and several measures of predictive accuracy were computed at the nucleotide, exon, and protein product levels. The data set used by Burset and Guigó was highly biased including only relatively short DNA sequences with simple gene structure and high coding density. Thus, the performance of the programs is likely to provide an overoptimistic estimation of the actual accuracy of the progrmas when confronted with large DNA sequences from randomly sequenced genomic regions of less standard composition and more complex structure.
Recently, a web site ( Banbury Benchmark site) has been established to serve as repository of long (100kb +) finished genomic sequences for which it is intended to provide a thorough experimental validation of all gene structures in the region (using e.g. cDNA selection/sequencing, exon trapping, promoter mapping and other methods of transcript mapping). These sequences will consitute a more realistic benchmark for computational gene identification programs in the large scale sequencing phase of the genome projects.
The evaluation by Burset and Guigó consisted of three steps:
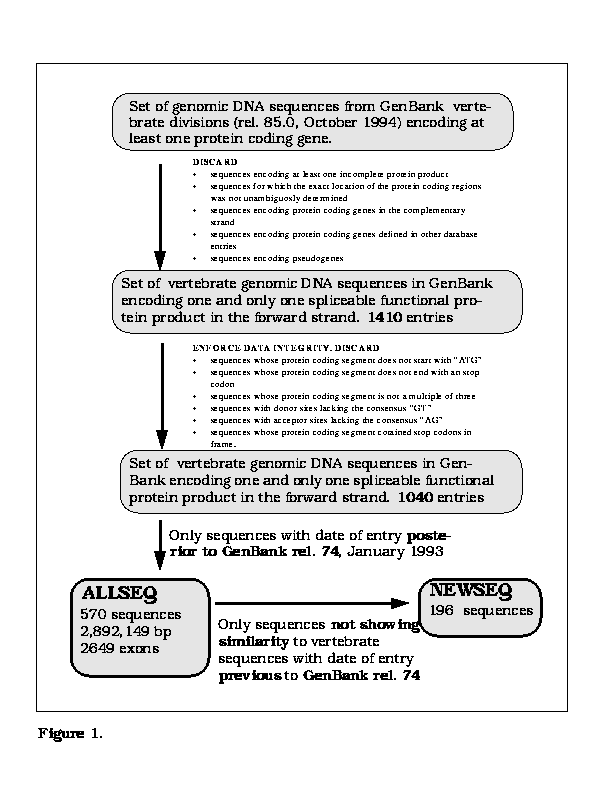
The ALLSEQ data set is characterized by two
features.
First, it includes only short sequences encoding a single
complete gene with simple structure, with high coding density,
and free of sequencing errors.
Second, ALLSEQ was built attempting to minimize the
overlap with the training sets used during the development of the
programs analyzed.
Thus, only sequences entered into the public databases
after January 1993 were selected into ALLSEQ. In addition,
a subset of ALLSEQ was considered
separately (NEWSEQ), comprising only those sequences that did not show a
significant similarity to the vertebrate sequences entered into the database
prior to January 1993. The ALLSEQ data set has 570 vertebrate sequences,
196 of them included in the NEWSEQ subset.
Files containing the DNA sequences in ALLSEQ, the locations of the exons in the sequences, and the corresponding encoded amino acid sequences can be found at /databases/genomics96/index.html
Prediction accuracy per base coding/non-coding.
Sn: Sensitivity = TP/(TP+FN)
TP:true positives
TN:true negatives
FP:false positives
FN:false negatives
Prediction accuracy with respect to exact prediction of exon start and end points
Sn: Sensitivity = fraction of correct exons over actual exonsPredcition accuracy with respect to the protein product encoded by the predicted gene.
% Sim: percentage similarity between the amino acid sequence encoded by the predicted gene and the amino acid sequence encoded by the actual gene


| Base-level | Exon-level | |||||||
|---|---|---|---|---|---|---|---|---|
| Method | Sn | Sp | AC | Sn | Sp | (Sn+Sp)/2 | ME | WE |
| FGENEH | 0.77 | 0.85 | 0.78 | 0.61 | 0.61 | 0.61 | 0.15 | 0.11 |
| GeneID | 0.63 | 0.81 | 0.67 | 0.44 | 0.45 | 0.45 | 0.28 | 0.24 |
| GeneParser2 | 0.66 | 0.79 | 0.66 | 0.35 | 0.39 | 0.37 | 0.29 | 0.17 |
| GenLang | 0.72 | 0.75 | 0.69 | 0.50 | 0.49 | 0.50 | 0.21 | 0.21 |
| GRAILII | 0.72 | 0.84 | 0.75 | 0.36 | 0.41 | 0.38 | 0.25 | 0.10 |
| SORFIND | 0.71 | 0.85 | 0.73 | 0.42 | 0.47 | 0.45 | 0.24 | 0.14 |
| Xpound | 0.61 | 0.82 | 0.68 | 0.15 | 0.17 | 0.16 | 0.32 | 0.13 |
| GeneID+ | 0.91 | 0.90 | 0.88 | 0.73 | 0.70 | 0.71 | 0.07 | 0.13 |
| GeneParser3 | 0.86 | 0.91 | 0.86 | 0.56 | 0.58 | 0.57 | 0.14 | 0.09 |
GeneID+ and GeneParser3 incorporate results from amino acid database searches to make the gene predictions.
The Banbury Benchmark site is intended to be a forum for scientists working in the field of gene identification and anonymous genomic sequence annotation, with the goal of improving current methods in the context of very large (in particular) vertebrate genomic sequences.
Different methods are based on different approaches (ab initio, training set dependent, statistical bias, motif/database similarity, etc ..), or often, combinations of approaches. For this reason, direct competition between them is not meaningful. The aim instead is to provide new sets of experimentally verified data for blind testing of the programs, so that others as well as the authors can see how they perform with respect to their own specific goals.
Roderic Guigo (i Serra), IMIM and UB. rguigo@indy.imim.es