In what follows, we revised the key concepts regarding the eukaryotic gene and genome structure needed to understand the annotation of genomes. In this context, annotation refers to the description and location of genes and other biologically relevant features on a genomic sequence. Our main goal is to fully comprehend what genome annotation projects offer and, as important, what they do not yet provide.
In this regard, it's worth noting that current gene prediction programs, among other bioinformatics tools, systematically ignore the complexity of eukaryotic gene structure. Diversity comes from alternatively spliced gene structures, non-canonical signals (that affect either splicing or translation) and from regions that control gene transcription, promoters, which are not yet well understood. Here, we give and overview to see some of the limitations and future directions in the gene prediction field.
Human chromosomes
It is time to introduce the three main genome browsers and check which genomes they are serving. In this practical, we will stick to the Ensembl server, but feel free to browse the other later on.
Questions:
Now, select human and make sure you understand main concepts in this page. Contrary to the view reported in newspapers, the genome sequence and its corresponding annotations is highly dynamic. There are two levels at which data can be updated: 1) sequence level; and 2) annotation level.
Questions:
-
The digital nature of the sequence (nucleotides: Adenine, Guanine, Cytosine and Thymine) permits an easy and symbolic computational representation as A, G, C and T letter codes, respectively. It is worth knowing that Uracil (U), which is in place of Thymine in RNA, is also written as T in sequence databases.
-
The double nature of the DNA helix, gives us two different sequences to analyze (with distinct encoded information). In order to handle such a dual data, the concept of forward or positive (+) and reverse or negative (-) strands and elements (genes, exons, introns...) is introduced. The forward strand, for us, is simply the original sequence we are working on. Note that this concept is meaningless in the cell, thus no differences are made between strands. For example, genes are transcribed from both chains.
-
The complementary nature of the two strands (A-T, G-C base-pairing), permits to work in the computer only with one strand (forward), the other (reverse) being conceptually retrieved when needed. Usually, genome projects only provide one sequence strand (forward) and forward and reverse elements are annotated on it, being the latter tagged as reverse when suitable. Usually, again, note that the cell has access, at the same time, to the two strands.
-
The anti-parallel nature of the double helix (due to 5' and 3' nucleotide ends), gives polarity to the strands. There is a general agreement to write DNA sequences from 5' to 3' (do not mix up this fact with the forward and reverse concept). The 5' region is also known as the upstream region and, therefore, the 3' region is also called the downstream region of the sequence.
- The triplet nature of the genetic code (a codon is 3 nucleotides long), permits to translate any potentially coding sequence in six different ways (frames). Three in the forward strand and 3 more in the reverse sense of the sequence. The finding of the right frame by the ribosome (it already knows the strand) is a challenging process to reproduce computationally.
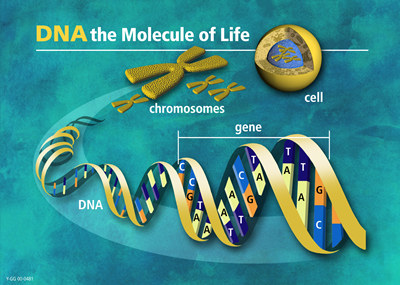
From DNA to chromosomes
Note that, in this course, we only analyze DNA at the primary sequence level. So, let's now check it. Please, select the chromosome 21 in the karyotype plot.
Questions:
Now, let's search for an specific gene over the whole genome. In the Find window, lookup the "alcohol dehydrogenase" gene.
Questions:
However, before analyzing the alcohol dehydrogenase gene, we will take an overview on the eukaryotic gene structure, processing and expression (see below).
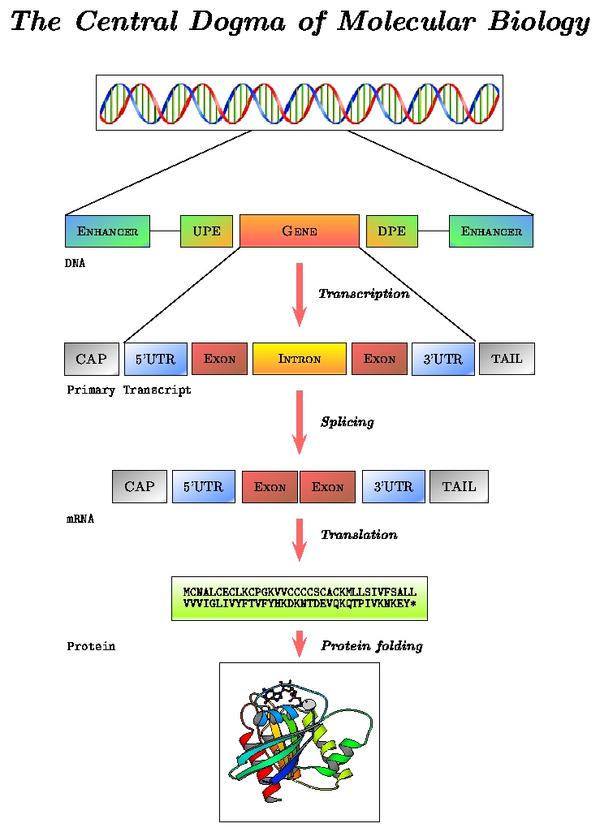
mRNA processing pathway
Locate in the alcohol dehydrogenase gene report the "Prediction Transcript" section. Note the correspondence between levels of reported data and processing steps:
- Exon information (gene structure on the DNA sequence): sequence before transcription
- Transcript information (mature mRNA sequence): sequence after splicing
- Protein information: sequence after translation
We will follow these links in the order shown, but first, move to the text below for a more precise discussion of the eukaryotic gene structure.
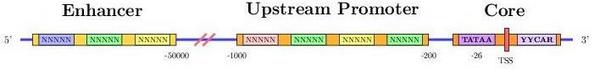
Promoter region
In short, transcription is the copying of DNA (template strand) to RNA (pre-mRNA). However, when analyzing mRNA, cDNA or EST data, bear in mind that the mRNA to be translated is, in sequence, identical to the coding strand (coding here always refers to translation, and not to transcription). That is, the mRNA is transcribed from the strand that has its complementary sequence. In conclusion, when annotating genomes, genes are annotated in relation to their coding strand.
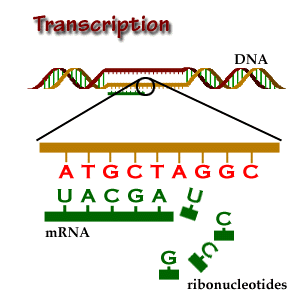
The copy of the template strand
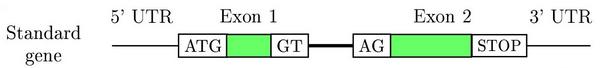
Schematic representation of a two exons eukaryotic gene on a DNA sequence
Gene prediction programs make use of this structure to find genes on a genome. Main characteristics are:
- Coding and non coding exons (UTRs)
- Introns
- Translation start site (ATG)
- Splice sites (GT, donor and AG, acceptor)
- Translation termination site (STOPs: TAG, TGA and TAA)
Questions:
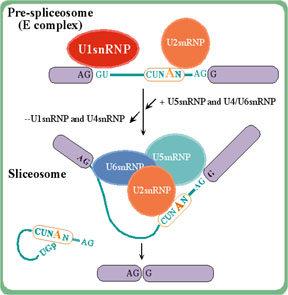
The spliceosome complex splices out
intron sequences
Follow the Transcript information link.
Questions:
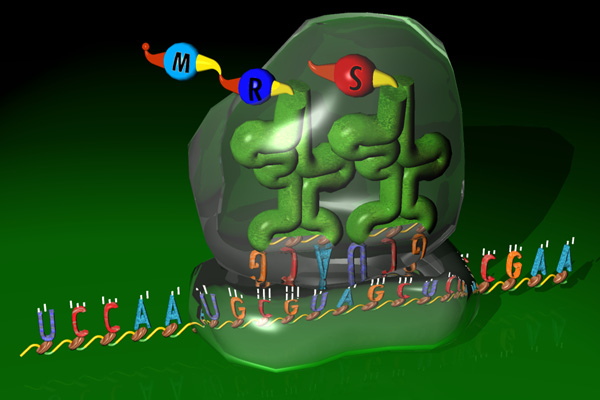
Translation maps RNA to proteins through a 3 to 1 letters code
On the other hand, there are three types of transcript data:
- mRNA: messenger RNA
- cDNA: a full-length (not always) and high-quality copy of an mRNA sequence
- EST: expressed sequence tag. A partial and low-quality copy of an mRNA sequence
Follow the Protein information link.
Questions:
Let's try now to get a more deep insight into the biological role of this gene. We will connect to a couple of web-based resources to learn more about our gene and protein. GeneCards is a database of human genes, their products and their involvement in diseases. Connect to this database and search for symbol/alias the approved HUGO adh symbol: AKR1A1 (as shown in the Ensembl gene report).
Questions:
Finally, we will try to get all the possible identifiers for this gene across several databases. Connect to GeneLynx and do a quick search in human for the HUGO ID: AKR1A1.
Questions:
We will browse the genomic region of the adh gene we are working with. Follow the this link. There are four levels of resolution:
In general, current gene prediction programs cannot predict alternative mRNAs in a reliable way, unless transcriptional data (mRNAs, cDNAs and ESTs) are available.
Data integration
It is about time to fully use the main feature of genome browsers: the ability to display all available information along a genomic region of interest. However, take first a look at the picture below to make sure you understand the pipeline behind these data.
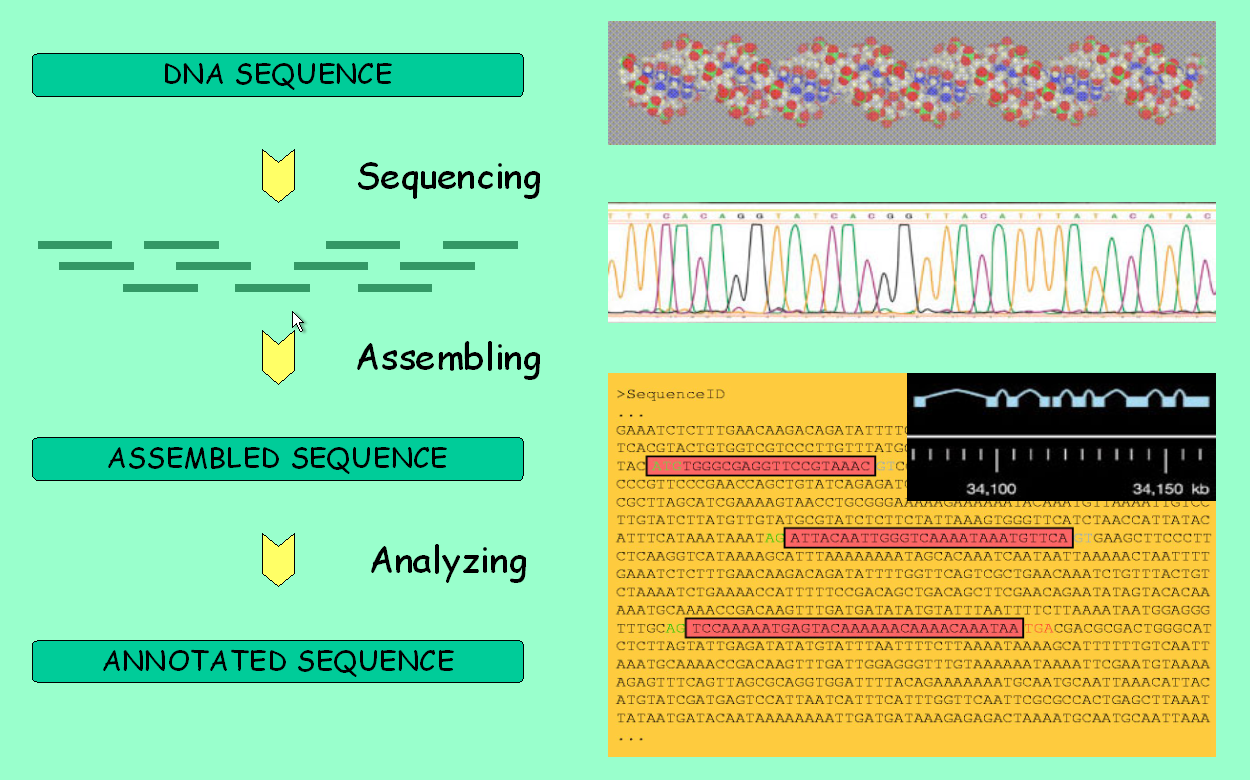
Genome analysis: from sequencing to annotation
Alternative Transcription
The transcription start site, can vary in the same gene depending on how the promoter region is activated. This results in a different pre-mRNA and, potentially, a differentially expressed mRNA or even a distinct protein may be achieved.
Alternative Splicing
Briefly, alternative splicing is an important cellular mechanism that leads to temporal and tissue specific expression of unique mRNA products. This is accomplished by the usage of alternative splice sites that results in the differential inclusion of RNA sequences (exons) in the mature mRNA.
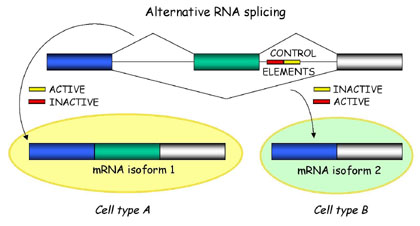
Alternative splicing produces unique mRNA products
Alternative Translation
Translation by the ribosome is a complex process. Source of variability are:
LINUX and genome annotation
Try to complete this practical.