grep '^ ' AC091491.embl | sed 's/[ 0-9]//g' > AC091491.fahowever, the fasta format requires each line to be preceeded by the '>seqname' line. We use emacs to introduce this line, and make the file AC091491.fa conform the fasta format.
Of course, there is a number of ways in which we can convert the original file AC091491.embl to the fasta formated one AC091491.fa, without the need of editing the file. Can you think of one such way?
It may be useful to have also the sequence in tabular format. The following script will convert the fasta file into a tabular file.
awk '{printf "%s", $0}' AC091491.fa > AC091491.tbl
Again, we need to use emacs to introduce a \tab between the
sequence name and the sequence itself, to get AC091491.tbl in the prober tabular
format. And, again a number of simple unix commands will do the whole
conversion automatically. Can you think on such command?
(Check here).
The tabular format allows to perform some preliminary analysis on the problem sequence
length of the sequence
awk '{print length($2)}' AC091491.tbl
152882
g+c content
awk '{print $2}' AC091491.tbl | fold -1 | sort | uniq -c | gawk '{print $2, $1/152882}'
a 0.323073
c 0.200802
g 0.189257
t 0.286868
RepeatMasker analysis produces a number of files. Among them:
By taking a look at this last file, we see that close to 40% of our sequence belong to repetitive DNA.It may be interesting to visualize the distribution of repeats along the sequence. There are a number of available tools to visualize genome annotations. We will use here gff2ps. There is a gff2ps web server at the pasteur institute, but the software can also be installed locally.
gff2ps requires the input file in gff format. The following awk script will do the conversion:
grep AC091491 AC091491.seq.out | awk 'BEGIN{OFS="\t"}{print $5, $11, "repeat", $6,$7,".", ".", "."}' > AC091491.seq.out.gff
Now we can run gff2ps: be careful about the path of the perl interpreter
(try which perl);
gff2ps AC091491.seq.out.gff > AC091491.seq.out.pswhich we can visualize using ghostview:
gv AC091491.seq.out.ps
gawk 'BEGIN{OFS="\t"}$2 ~ /Term|Intr|Init/ {print "AC091491", "genscan", $2, start=($4<$5 ? $4 : $5), end=($5<$4 ? $4 : $5),$13,$3,$7, $1}' AC091491.genscan | sed 's/\.[0-9][0-9]$//' > AC091491.genscan.gff
Now we can run gff2ps and visualize the two predictions
simultaniously:
gff2ps AC091491.geneid.gff AC091491.genscan.gff > AC091491.genepredictions.ps
After running the blast search, we store the results in, for instance, the file AC091491.blast.est
These are difficult to analyze. We will first visualize them using gff2ps. This requires converting the blast result to gff. It is possible to write a relatively easy perl script to do the conversion. Here, we will borrow one such script written by Josep F. Abril. The name of the script is parseblast. Then, we just type
parseblast -Gi AC091491.blast.est | gawk 'BEGIN{OFS="\t"}{$2=$9;$6=$7=".";print}' > AC091491.blast.est.gff
And now we are ready to plot the est matches along the query sequence:
gff2ps AC091491.blast.est.gff > AC091491.blast.est.psAs you can see, there is a large number of singleton EST matches. Because artefacts during EST library construction are not uncommon, usually only spliced ESTs are considered good evidence for the existence of a gene, when identified along a genomic sequence.
Let's consider, thus, only the spliced EST matches. To obtain them, we just need to identify those hps's appearing more than once in the gff formatted est search file. You can write a simple perl script for this. I wrote an awk script (save it in a file: getsplicedhsp.awk), instead, and named the resulting gff file of splices est matches, as AC091491.blast.est.spliced.gff
gawk -f getsplicedhsp.awk AC091491.blast.est.gff > AC091491.blast.est.spliced.gff
Now we can visualize est matches altogether with gene predictions
gff2ps AC091491.blast.est.spliced.gff AC091491.geneid.gff AC091491.genscan.gff > AC091491.est+genepredictions.psWe can convert the postscript output to gif (or jpeg) format, and include it within the html document.
convert -antialias -rotate 90 AC091491.est+genepredictions.ps AC091491.est+genepredictions.gif
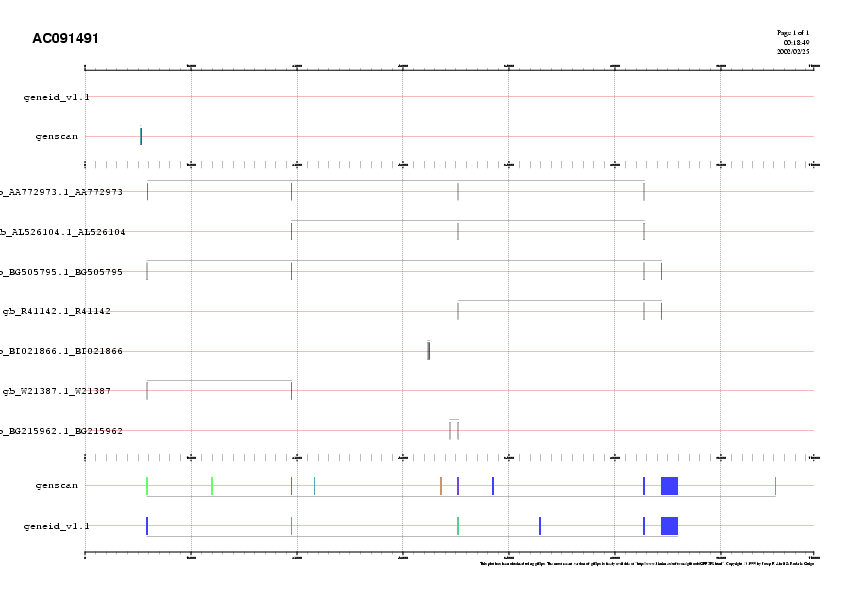
After looking at the plot, it appears, in this case, that the geneid prediction is quite consistent with the EST matches, with the exception of exon 68656-68712. genscan appears to predict a number of additional exons. Note, however, that the large exon predicted both by genscan and geneid is only partially supported by EST matches. In general, this does not necessarily means that the exon predicted by the "ab initio" gene prediction is incorrect. Because of the mechanism by means of which ESTs are obtained, 3' ESTs do generally support the 3' end of a gene, while 5'ESTs are often internal to the gene, and do not cover its 5' end. The ESTs matching our query sequence are
>gb|AA772973.1|AA772973 af33h09.s1 Soares_total_fetus_Nb2HF8_9w Homo sapiens cDNA clone
IMAGE:1033505 3' similar to TR:Q15111 Q15111 PHOSPHOLIPASE C. ;.
Length = 471
>emb|AL526158.1|AL526158 AL526158 LTI_NFL003_NBC3 Homo sapiens cDNA clone CS0DC015YO17 5 prime.
Length = 926
>gb|BG505795.1|BG505795 601860281F2 NIH_MGC_61 Homo sapiens cDNA clone IMAGE:4072604 5'.
Length = 630
>gb|R41142.1|R41142 Hk907-f Adult heart, Clontech Homo sapiens cDNA clone k907-f.
Length = 355
>gb|W21387.1|W21387 zb59h03.r1 Soares_fetal_lung_NbHL19W Homo sapiens cDNA clone
IMAGE:307925 5'.
Length = 254
Because supported by a 3'EST (AA772973), we can we quite confident of the 3' end of the gene. However, despite a number of 5'ESTs, we can not guarantee the 5'end of the gene, which could extend quite beyond this very large exon (as, both, the genscan and the geneid prediction suggest)
Thus, we conclude that a likely exonic structure for the (apparently) only gene in contig AC091491
is
AC091491 prediction Terminal 9279 9458 . - 0 AC091491 prediction Internal 31099 31235 . - 2 AC091491 prediction Internal 56242 56317 . - 0 AC091491 prediction Internal 84280 84483 . - 0 AC091491 prediction Internal 87031 89517 . - 0There is one EST (BG505795) suggesting an alternative form for this gene, in which the short exon 56242-56317 is missing. There are two spliced ESTs (BI021866 and W21387) wich support exonic structure incompatible with the exonic structure supported by most ESTs and predicted by the "ab initio" gene prediction programs, and are deemed as artefactual.